Myelodysplasia
(MDS)
Click for more info on MDS Syndrome
General Discussion
Myelodysplastic syndromes (MDS) are a rare group of blood disorders that occur as a result of
improper development of blood cells within the bone marrow. The three main types of blood cells
(i.e., red blood cells, white blood cells and platelets) are affected. Red blood cells deliver
oxygen to the body, white blood cells help fight infections, and platelets assist in clotting to
stop blood loss. These improperly developed blood cells fail to develop normally and enter the
bloodstream. As a result, individuals with MDS have abnormally low blood cell levels (low blood
counts). General symptoms associated with MDS include fatigue, dizziness, weakness, bruising and
bleeding, frequent infections, and headaches. In some cases, MDS may progress to life-threatening
failure of the bone marrow or develop into an acute leukemia. The exact cause of MDS is unknown.
There are no certain environmental risk factors.
Myelodysplasia (MDS) is sufficiently uncommon that its incidence is only reckoned to be between 0.5 and 10 people per 100,000 of the population. It is this rarity which makes it so difficult when trying to find information about it, for it is not even considered worthy of mention in most of the normal family or popular medical books available to the public. The normal general practitioner very seldom comes across a case and he will have enough difficulty keeping up with the rapid advances in the illnesses he is most likely to meet without spending too much time mugging up on such a rare one, where a patient suffering from it may never, or only very seldom, cross his threshold. Myelodysplastic Syndrome is a bit of a mouthful, so for convenience is normally shortened to MDS. Why the name? Well, myelo = to do with bone marrow; dysplasia = the proliferation of cells of an abnormal type.
Essentially this is a malfunction of the bone marrow - the material in the hollow centre of bones which produces and refreshes the various cells which make up the blood. It does not stop production altogether but gradually produces less cells and those which are produced are often misshapen or don't properly mature and so are unable to carry out their normal function. Gradually you lose the benefits which each type of missing cell brings. So initially we just need to refresh our knowledge a bit about blood .......
First, stem cells are produced in the bone marrow. These divide again and again to manufacture both more of themselves as well as blast cells. These latter are immature cells which in turn differentiate to produce the mature red, white and platelet cells as required.
Red cells carry hemoglobin around the body and it is this which transports oxygen to the muscles, brain, heart and so on and effectively provides the sustenance to keep them active. It also carries the waste carbon dioxide back to the lungs to be exhaled and lost. Reduce this effect and you soon get physically tired and additionally start gasping for breath as the body calls for more oxygen and your lungs try to cope with the demand. These are the longest-lived cells at about 120 days. After this, they are broken down to bilirobin by the spleen and leave the body as bile discharged by the liver to the intestines. (There is no need to remember this last bit; suffice to say that at about 120 days' age they are pushed out of your body and lost). These, of course, are the cells which give blood its red color. Reduce them, as in Myelodysplasia, and the blood takes on a thinner character and is less intensely red.
White cells provide the essential defense against infection, attacking and destroying invading forces such as bacteria and viruses. They also remove dead or injured cells from the body. They come in various types (beyond the scope of this account) and their life is very short at just about 20 - 30 hours.
Platelets are the third main cell type. These are the smallest of the lot and serve to coagulate blood and so stop bleeding at any wound. They have a life of around 8 to 12 days. All of these cells are carried around the body's circulation system in a clear protein-rich liquid called plasma.
Right: that's it for blood, but we shall return to it later - can't get away from it when considering MDS.
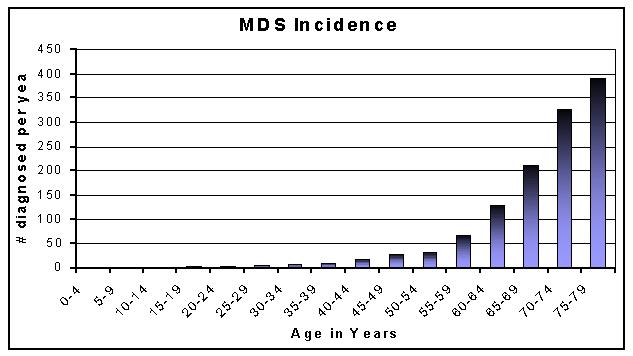
Age is a dominant factor. Nearly 30% of sufferers are between ages 60 & 70 and nearly 60% between 70 & 80. It is pretty rare in young people. Interestingly, up to age 50 slightly more women than men have it: after 50, men are preponderant with the disparity increasing with age. So maybe there is a hormonal effect partly at work. [The graph below is based on figures from a Leeds University study of 1200 patients carried out under the auspices of the UK Leukaemia Research Fund]
Well at the start, most people don't know - it tends to be symptom less. A normal way of finding out is by chance when blood is tested as part of a routine medical check-up or, say, tested in advance of an operation for some other cause. Generally, the disease is progressive; as it progresses, symptoms of anemia will start to show themselves and testing for this could indicate that the cause is MDS rather than normal anemia.
If this is the case, then a bone marrow biopsy would be carried out. Sounds awful but in practice it is usually OK (see here for a discussion of BMBs). Under a short anesthetic a small sample of your bone marrow is taken out (normally from your large hipbone) and also a fragment of the bone itself for examination under a high-power microscope. This can show whether the primary cells in the bone marrow (the stem cells) from which the others derive are in good order or not. In this way, it can not only be determined whether you have MDS but also which variety (see below for varieties). Examination can also detect any chromosomal abnormalities, one of the possible causes of the disease, and important in determining likely outcomes.
No, neither does it seem to be hereditary.
Really the cause is still unknown. There does seem to be a fairly well-established connection with a long exposure to benzene or following high doses of radiation or chemotherapy (one study in Denmark has even suggested that aircrew on long-distance, high-altitude flights may be more susceptible than folks on terra firma due to cosmic radiation!); but I would guess that none of these has been a factor with the normal patient. I have read of the possibility of inhalation of some types of spores from black mould or of perhaps a connection with some types of insecticidal sprays. It may possibly arise from a mutation in one of the genes of the primary stem cells in the marrow.
But reading the literature, still nobody seems to have a finger on a definite cause. Maybe there is more than one. Maybe it is just that in most cases your bone marrow gets tired as you get older and so acts in an enfeebled way. The term given to such doubt by the medical profession is idiopathic, which loosely means "haven't a clue".
Normally, yes, though the length of time it takes to happen is very variable. It is not the MDS itself which kills but the downstream illnesses which it can engender. Statistically, some varieties of MDS allow the patient several years; others, I am afraid, run for only a few months. See details below under prognosis.
There seems only one fairly sure treatment offering a cure. This is by bone marrow or stem cell transplant (BMT/SCT). However, like any transplant, your body tries to reject anything foreign introduced into it, resulting in what's known as Graft vs. Host Disease (GvHD). To minimize this effect, treatment is only considered if a compatible donor can be found to provide healthy replacement bone marrow or stem cells which will not conflict with the patient.
So, not surprisingly perhaps, the best source is from a brother or sister; siblings are the most likely to have that perfect match. Even with them, though, there is no guarantee of compatibility. There can be others whose marrow might suit and these days national registers are compiled of people willing to donate some of their bone marrow to help others (and I'd strongly encourage people to register if they are of suitable age!). If a match is found from this source, you are in luck.
Transplants can be either bone marrow or stem cell. If they use bone marrow, actual marrow is taken from the donor's hips and infused into the recipient. If they use stem cells, the donor is hooked up to what's known as a pheresis machine. This takes blood from one arm, separates out the stem cells, and returns what's left to the donor's other arm. The stem cells are then infused into the recipient.
There are advantages and disadvantages to both. There is frequently somewhat less Graft vs. Host Disease (GvHD) with marrow. Stem cells usually engraft more quickly, leaving the recipient less time to be vulnerable to infection or bleeding before counts return to normal. Marrow transplants have been done for over 30 years. Stem cells transplants have been done only for the last few years and only recently have they begun using stem cells from unrelated donors.
However, it is a major undertaking, sufficiently so that it is normally only carried out on those under age 55 because of the severity (see below). However, upper limits range from 35 to 70, depending on the nature of the BMT and the quality of the donated material. Get that wrong, and you become more likely to die of the treatment rather than the disease. Not the intention at all!
It involves killing off the existing cells by means of whole-body radiation and/or chemotherapy carried out over several days This kills them all, good and bad, to avoid existing cells attacking the newcomers - the graft vs. host disease mentioned above. Inevitably with all the white cells gone, the body has no defense against invading diseases, so the surrounding area must be completely sterile and kept so throughout the several weeks the patient is in hospital. It is a dire and strongly traumatic operation and it must be faced that there is a fairly strong possibility of death, at the time or even up to a year afterwards. Not so bad as it was in the early days of BMT but still a factor.
There are three main types of transplant. Autologous receive their own stem cells/marrow, taken out and put back in later. Syngenetic comes from an identical twin. Allogeneic means derived from a remote source, i.e. other than the first two. (The allo part of the word is not, as it sounds, a Cockney greeting but comes from the Greek allos = afar).
Another difficulty with BMTs is that they are expensive - one quoted cost for a US Centre was in the region of USD 0.25M, and although I have no similar figure for costs elsewhere, it is an intensive and costly exercise. This inevitably brings up questions of insurance coverage in systems where availability of treatment depends on availability of payment. Medicare, for example, does not cover this procedure until an acute leukemia develops. Catch-22 ....
"Surely there must be a better way than this" went the feeling. It turned out there was. About four years ago, research came up with the mini-transplant, a process which is still looked upon as under development but has been carried out in several countries around the world. It still requires donor marrow but the advantage of this technique lies in the much lower dose of chemotherapy &/or radiation therapy required in the pre-transplant stage; the minimum of dosage, hence the name.
So straight away this is much less stressful on the body than the normal BMT. A mini-transplant's lowered treatment regime does not necessarily immediately destroy all the patient's rogue cells but cells in the transplanted marrow apparently recognize the existing aberrant cells and gradually set about destroying them. Both lots of cells present at once is called mixed chimerism - luckily you don't have to remember this name. Prevention of rejection can be achieved by giving small doses of the drug fludarabin before transfusion, or newer drugs such as orBec (Beclomethasone Dipropionate). Such transplants allow later infusion of white blood cells (T-lymphocytes) from the bone marrow of the original donor where this will help in topping-up the process.
The result is that instead of several weeks in hospital for the normal BMT, the procedure can be carried out within a day at an out-patients' clinic. The nausea/diarrhoea met with in conventional treatment are now minimal and hair-loss avoided. All this must surely be an enormous advantage. Additionally, since it is so much less severe, it can seemingly be carried out on patients even in their 60s and 70s - well beyond the normal BMT cut-off age.
Up to the time of writing, hospitals have been using sibling-matched marrow as the obvious safer route to start the new process but are gradually extending the range to include non-family marrow-bank donors. At the moment, this mini-BMT is only available at relatively few hospitals: I gather that in the USA, in autumn 2000 there were only 23 offering the technique as opposed to around 115 offering conventional BMT. There are two in Canada (Montreal and Quebec) and several in UK (eg Kings College Hospital, London). But the advantages seem so great that surely there must be a rapid spread.
I must stress that I know little of this technique other than what is stated here. It has been in semi-experimental operation for only a few years and so it is too early yet to know how long remissions will last and whether there will be late-arriving graft vs. host problems. If you are interested, you or your physician should check out Web pages on the subject. Key-in mini-transplant on one of the search engines (Google) and see what turns up.
A recent development is that the level of matching required for a BMT may in certain circumstances not be as tight as previously thought. In a study at the Fred Hutchinson Cancer Research Center (results will appear in the Dec. 20 2001 issue of the New England Journal of Medicine - but you'll have to subscribe or pay for the article if you want to see it!), scientists discovered that some tissue-type mismatches are permissible in bone-marrow or stem-cell transplantation to treat leukemia. This knowledge opens doors for many people who are unable to find fully-matched donors - including perhaps MDS sufferers.
I have included some links to more information on transplants that are written in a fairly comprehensible style on the 'Useful Links' mini-topic page.
If you are not a candidate for one or other bone marrow transplant - and as explained, by the nature of the disease, most people are not because they are probably in an upper age group or cannot find an acceptable donor - then the standard treatment is via some of the new drugs being developed or what the doctors call supportive or palliative care.
This latter amounts to periodical transfusions of red blood cells (and sometimes platelets) which have been isolated from donated whole blood. Matches here are much easier than with bone marrow transplant and though inevitably nobody really wants to have someone else's blood put into them, these days blood is so carefully screened that the chance of getting some 'nasties' in the process is pretty slight. Better to take that minuscule risk than to die any rate. However this is not a cure; it is only an alleviation of the symptoms of severe anemia.
The transfusions need to be carried out whenever the patient's number of red cells falls too low and this point will depend on a particular patient's point in the passage of the disease. This determines how many units of blood are needed and how frequently they need be given. Typically, for men, a red cell count of 9 g/dL is about the point at which severe breathlessness sets in and the need for transfusion probably arises every few weeks. Women seem to run about 2 points less (for comparison, a normal level for a healthy man would lie in the range 13.5 to 18 g/dL).
The reason for this frequency of transfusions is that fact about cells which was mentioned earlier, that they have a limited life. It would be nice to think that the 120 days' life of red cells meant that that was as frequently as transfusions were needed. But the blood you get will contain a mixture of cells of different ages; some will be recently formed, others will be close to their 120 day 'sell-by' date. It is this fact which results in transfusions being needed every few weeks.
As an example, I get either two or three units (a little under 1 pint each) every four weeks or so. That is insufficient to relieve me of the symptoms of breathlessness completely but it does ease them. You soon get used to the transfusions; the chief trouble with them is the sheer boredom of sitting there for several hours as the donated blood is slowly dripped in. Take a good book to pass the time. I find that the transfusion takes about 36 hours before the benefit becomes apparent.
To determine the timing of transfusion need, a small blood sample is taken from you regularly for analysis, both for the actual cell count and also for compatibility testing for any donated blood. Nowadays this analysis is carried out automatically by machine and the result printed out very quickly. I can recommend it as good practice to obtain these figures from the laboratory producing them and to plot them on a graph each time. This way you can follow the progress of your MDS, though it must be admitted that to watch the cell counts dropping month by month is a bit depressing. Still, it is good to know just where you stand. Plot HGB, WCC, PLT and possibly Neutrophils - all these should be on the printout.
One downside of transfusions is that over time you acquire more iron in your system from each unit you receive, stored in and around major organs in the form of a compound called ferritin. Iron carried around the body by hemoglobin is not lost but recycled when fresh cells are produced. If you are on red cell transfusions, you will be adding small amounts of iron to your body's existing stock each time you receive blood. Over a length of time this can build up to produce what is called iron overload (or haemochromatosis to give it its Sunday name), it is wise not to add yet more iron by way of tablets.
Fortunately, should overload occur, this can be corrected by giving a drug named deferoxamine (Trade:'Desferal') which serves to remove the iron from the body. There are two methods of giving this drug: see this extra bit if you're interested. There is another drug, Deferiprone, which has been used for the same purpose of iron removal for many years in the treatment of sickle cell anemia. This is taken orally in tablet form and has fairly recently received approval from the European drug regulatory body (but with 'contra-indications' for neutropaenia - that's a shortage of one of the white blood cell types to you & me - and that probably rules it out for many MDS sufferers). Also, one study has alleged that there are serious side effects associated with Deferiprone, mainly liver toxicity and scarring.
Over time, this can cause damage in its own right, and needs to be reduced. There are two drugs used for this purpose -
There are some other treatments which are worth considering.
Unfortunately it is very expensive, is not a permanent cure (the patient drops back later if it is stopped) and only works at all in under one-third of MDS patients. Still, if you can afford it, don't mind injecting it and are willing to take the chance that you may fall into the two-thirds for whom it does not work, then it is worth a try. For me, a three months' trial (including G-CSF - see below) was an expensive failure.
Folic Acid, Vitamins B6 & B 12. I have seen these mentioned as worth a try. They are obtainable over the counter without prescription in chemists' shops, since they often appear as 100% of Recommended Daily Allowance when they are constituents in multivitamin tablets. The consensus of opinion, however, seems to be that they are useless for MDS alleviation used alone, though research is in progress to combine them with weak chemotherapy in a treatment. Early days yet to know whether it will work.
If you should try them, be sure by checking the table of contents on the package not to get the type containing iron as one of the constituents - see above about iron overload.
All patients treated with 140mg or above of the drug showed a marked decrease of the proliferation of white cells characteristic of the disease. But at the 300mg dose rate, 23 out of 24 (96%) had normal cell counts. So great was the success that word flashed around the world via medical conferences and, inevitably these days, via the Internet. Just as inevitably this prompted a hullabaloo from those suffering from CML "Hurry up that testing; we're dying out here!" So an accelerated programme was put in place. The trade name following approval from the US Food and Drug Administration (FDA) in May 2001 is Glivec ?. Approval was granted as 'Orphan drug' status in Europe in November 2001, although it will not receive UK NICE approval until August 2002.
However the most likely progression for MDS patients is to acute myeloid Leukaemia (AML), not CML. Tests of the drug are now underway to assess its effectiveness against AML as well but are not so far advanced as against CML at the time of writing. The standard present treatment for CML is Interferon, but results seem to be patchy and require daily injections. STI-571 is a pill taken orally and in the main has only few, pretty mild, side effects. It is too early to say how long remission will be achieved and also whether patients will have to live permanently on the new drug. In particular, recent studies have started to look at Glivec in combination with compounds such as arsenic trioxide.
There's a bit more on this drug on this 'mini-topic' page.
There seems to be no universal response, either; where some people have their MDS symptoms improved with a trial drug, others show no improvement and often any improvement is very slow, measured in months rather than days or weeks. You feel it must be a maddening challenge for the researchers.
To get on to one of these research regimes, it would be necessary for your physician to make the contact with the company producing the drug or the research body. But which one? How can your doctor sort the wheat from the chaff, make his way through the maze of trials when there are so many and so much of the technical stuff is so abstruse? The only way I can see is for him to know somebody engaged in the art and take advice from them. It doesn't hurt to do a little check around on the Internet for oneself as well. Quite a few of the results get published there, but I warn you - it's heavy going!I cannot advise, but three drugs which correspondents or literature have said have shown promise are Amifostine, Azacitadine and Thalidomide. However I only mention the names: I have no experience of the qualities of them. The Thalidomide, incidentally, is the same drug that 30 years ago produced babies without limbs when given to pregnant mothers, now resuscitated and showing significant promise in treating MDS. It seems its main role is in boosting production of red blood cells, thus reducing the need for transfusions, but there is some evidence that its effect wanes after about a year of use.
Really, though, this is far too complex a subject for this article. Just to quote a few of the drug names under test gives an idea of the research range - Cyclosphosphamide, Fludarabin, Bisulfan, Antithymocyte Globulin, Dolastatin 10, Decitabine, Rubitican or the unlikely-named flt3L ......... so the list goes on. My favourite is TOPOTECAN. Does that not sound like the name of one of those lightly-discovered third-world nations? You can almost hear the news announcer - "Today our correspondent in Topotecan has reported an attempted coup against the existing government ........"! Should you wish to check further on trials in progress, try www.ClincalTrials.gov, and when asked, key in 'Myelodysplasia'. This brings up details of about 50 trials of various drugs. Alternatively, go to A.A and MDS International's home page for Patient's Clinical Trials.
No good beating about the bush - this disease is a killer if you are too old for bone marrow transplant and maybe even then. The only two questions are 'how long will it take?' and 'what will I die of?' But the inevitability need not be too depressing - after all, we all have to die sometime. As the old saying has it, "The only certain things in life are death and taxes!"
Look at it this way ....... those of us with MDS have an advantage over someone incurring a sudden death, say, in an accident or from a heart attack. They die with no chance of putting their affairs in order, saying goodbye to loved ones or generally tidying up their life's end. We are given that warning some months, or even years, ahead. So we must make good use of the time available to us.
Make sure that your will is in order and bring it up to date if necessary (when did you last look at it?), see that any dependents left behind have their financial position safeguarded, look up those friends you have meant to see for a couple of years and never quite made it, perhaps take that short cruise you always meant to go on or visit some area you have always cherished for a last look ....... that sort of thing. But be careful to work within the limits of your health. You are probably going to be pretty restricted in what you can do because of the breathlessness and the lethargy that goes with anaemia. It makes sense to do anything moderately active immediately after you have had a transfusion to pump up your red cells.
This depends upon the variety of MDS you have and how far advanced you are in that variety. In 1982, haematologists from France, America & Britain got together to devise a classification for the varieties of MDS, which, taking the first initials of their countries they called the FAB system. I know the inevitable reaction of most folk will be to say "that's more than I want to know on the subject"; nevertheless knowing about this classification is vitally important. So, please, stay with me and give it a go - it's not all that bad. Starting from the least vicious to the worst, these are:-
1 Refractory Anaemia (RA). Refractory means not responsive to treatment for anaemia, which would normally be iron or vitamin additions. It is characterised by only a small percentage of blasts in the marrow, under 5%. Survival is good, averaging about 4 years or so and only around 10% move on to Leukaemia.
2. Refractory Anaemia with ringed sideroblasts (RARS). These are red cells that can be seen under the microscope to contain iron, normally concentrated a little inside the edge of the cell. Under 5% of the marrow cells are blasts. Survival average is 4 to 5 years and under 5% move on to leukaemia.
3. Refractory Anaemia with excess blasts (RAEB to avoid a mouthful). 5 to 20% of the marrow cells are blasts (i.e. not developing into useful cells) and up to 5% of these blasts may escape to circulate uselessly in the blood. Average survival is 2 years but despite these abnormalities, progression to leukaemia is only 20 - 30%.
4. Refractory Anaemia with excess blasts in transformation (RAEB-t.) Here many more blasts are present in the marrow, 20-30%, and more than 5% circulate in the blood. Now things are very serious. Survival is only averaging at around 6 months and 75% who reach this stage go on to develop leukaemia. Not nice.
Unfortunately, deciding which type you represent in the scheme of things is not susceptible to a
DIY approach. It needs the blood and marrow checking with a microscope of several hundred times
magnification - which, let's face it, few of us own. So you are in the hands of the specialists.
Don't treat those average expected life spans as gospel, though. Opinions vary quite a bit between
various research bodies and they are in any case only averages. To draw on my own experience, I have
RAEB and of the reputed 2 years life, I have now had nearly 3 (was 3.5 at the end - IMB) and
am still above ground and mobile - pretty breathless sometimes, yes, but otherwise refuting the
statistics. Additionally several of the life expectancy figures were produced some years ago before
mini-transplants or some of the newer drug regimes. These must serve to raise the prognosis figures,
so they probably need revision.
Nothing in life is simple. That FAB system, which has served for the best part of two decades, has now been partly superseded by the International Prognostic Scoring System (IPSS). I won't go far into this except to say that it brings into the calculations of life expectancy the factor of chromosomal irregularities, as well as blood count and blast percentages as in the FAB system. Each factor is awarded points, depending upon its seriousness (rather like those magazine questionnaires that ask questions, allot marks for answers and from the total, tell you whether or not you are the perfect lover!). When these are totted up, they can be referred to a table that gives a prognosis for that total. Just a refining of the old order but that chromosomal factor is very important. If you want to know more about the IPSS, look here or here. Not only that, but the World Health Organisation has proposed another classification, although some of the haematologists working in the field find this one a bit problematic from a prognostic or clinical perspective. There's also a useful comparison guide between the three systems on the American Society of Hematology education site.
MDS is not in itself a killer but the ailments in induces can be. It is convenient to relate these to the deficient blood cell types.
Lack of red cells, as mentioned earlier, makes you very short of breath and is generally debilitating. As the saying has it," my get-up-&-go has got-up-&-gone". This is common to all forms of anaemia. It also helps to induce or accentuate lung trouble, which can lead to acute bronchitis or pneumonia, both potential killers. There is also strain put on the heart, since it beats more rapidly to try to make up for the oxygen deficiency consequent upon the low red cell numbers. Eventually it could give up the fight and fail.
Lack of white cells means that your resistance to bacterial and viral infection is reduced. These, after all, are the body's main line of defence. So you will be pretty wide open to attack by a range of nasties, some of which, like respiratory ailments deriving from influenza, or infection getting into wounds and then spreading, can turn out to be fatal if not checked. In view of the very short life of white cells, this is difficult to guard against.
Low platelet count can result in spontaneous bleeding, so bleeding from the gums or nose is not uncommon. These are not much of a worry but internal bleeding from any of several organs certainly is.
For some, the final illness will be acute myeloid leukaemia (AML). If the MDS has progressed through the various stages of the disease mentioned in the previous paragraph, then this will be the ultimate. Those blast cells no longer differentiate into the three main blood elements and so, unaltered, they start to proliferate in the blood. This makes the blood light-coloured, hence the Leuk- bit of the word (Greek leukos = white). MDS used to be called pre-leukaemia or smoking leukaemia before the advent of the FAB classification and that is about right.
Here is a list of somewhat disconnected points which it strikes me are probably worth mentioning:-
1992-2025 DC2NET, Inc. All Rights Reserved
| Software | Text Links | Sciatica Back Pain | Web Hosting |